What We Offer
Genetic Counseling/Consultation
Our geneticists/genetic counselors obtain detailed family histories, analyze inheritance patterns, identify at-risk individuals, and help prospective parents to understand their particular genetic risks, options, and likely outcomes in pregnancy. We explain the often complex world of new genetic tests and help patients make important decisions about their pregnancies. Changes in technology have occurred so rapidly that most primary obstetricians do not have the time to be able to explain these issues to their patients in an effective manner. Today, many hospital systems have their genetic counselors either supplied by laboratories – who have mixed loyalties – or within pediatrics departments for whom prenatal is only a small part of their portfolio.
Prenatal issues are what we do every day.

The enhanced genetic screening (EGS) program is often used as the framework in which many of our services are provided, though our services are also offered separately. What to expect in the EGS program:
- For most patients, particularly those under 30, the odds of having a child with a Mendelian disorder (such as cystic fibrosis, SMA, or one of hundreds of rare disorders) is actually HIGHER than having a baby with Down syndrome or other commonly recognized chromosome conditions.
- Such screening allows us to identify “at risk” couples. In the past, most of the time we would perform prenatal diagnosis for such conditions, it was because the couple already had an affected child. Now we can analyze BEFORE the couple has the life altering circumstance of an affected/special needs child.
- We obtain and analyze a couple's family history, personal history, ethnic groups, medications, and other information, to determine if there are any risk factors for genetic abnormalities above the average 3% population risk of having a baby with a birth defect.
- Even for couples who are in known at risk groups (e.g., Tay Sachs disease and Ashkenazi panel) when they do have a child affected with this type of disorder (Mendelian), more often than not, the disorders does NOT fall within the Ashkenazi Panel.
- To identify carrier status for genetic disorders, blood-screening tests are offered. The model for screening has changed in the past few years as expensive, ethnic-group-specific panels have been almost completely replaced by a "pan ethnic" (i.e. everyone) screening for dozens to hundreds of disorders regardless of background. New technological approaches now make it cost effective to offer a large panel of screening tests to everyone regardless of the couple's ethnic group or groups. As the data have accumulated, it is very common to uncover that many patients that their actual ethnic background is not what they presumed.
- New technological approaches now make it cost effective to offer a large panel of screening tests to everyone regardless of the couple's ethnic group or groups. As the data have accumulated, it is very common to uncover that many patients that their actual ethnic background is not what they presumed.
- For genetic counseling we still specify that the likelihood of being a carrier of certain diseases does vary tremendously by background. For example, for patients of Ashkenazi Jewish descent, there has been a continually increasing panel of disorders including well-known conditions such as Tay-Sachs disease and many others just as common but less well known. Patients of African heritage are tested for sickle-cell anemia. Patients of Mediterranean origin are at high risk particularly for β-thalassemia. Patients of Asian descent are at high risk for α-thalassemia. Many other carrier screens are possible but had not been routinely performed because of low incidence and high expense, but now with pan ethnic screening these can be investigated in a cost-effective manner.
- Many other carrier screens are possible but had not been routinely performed because of low incidence and high expense, but now with pan ethnic screening these can be investigated in a cost-effective manner.
- To clarify the risks for Down syndrome and other chromosomal abnormalities, screening via first-trimester ultrasound, including nuchal translucency measurements, is performed in conjunction with blood tests.
- Microarray lab testing shows the detection of serious anomalies caused by small gains or losses in chromosomal material in over 1% of cases which is comparable numerically to a 38 year old for common problems like Down syndrome. However, such testing can only be done on fetal tissue (e.g. obtained by CVS or amniocentesis) specimens. Therefore, we offer prenatal diagnostic tests to all patients regardless of their age.
- Chorionic villus sampling (CVS) in the first trimester, and amniocentesis in the second, are used to determine chromosomal status, such as in Down syndrome and now hundreds of other disorders. Other DNA tests are used for disorders such as cystic fibrosis, Tay-Sachs, or sickle-cell anemia.
Chromosomes and Screening
Screening vs. Testing: There are two different types of tests. Understanding the difference is very important.
- "Diagnostic" tests, such as CVS and amniocentesis, are meant to give a definitive answer to a specific question. They are generally Yes/No. These tests are sometimes expensive and may also have risks associated with being performed. They are commonly only performed on patients known to be at high risk.
- "Screening" tests are those usually offered to everyone and are intended to alter the odds of there being a problem. They do not give a definitive answer but can help patients decide if they wish to pursue diagnostic test to get the definitive answer.
- All pregnancies have a risk of chromosome abnormalities such as Down syndrome (DS). While many patients are most concerned about DS because that is the most commonly discussed condition, the reality is that it is only one of a large number of conditions that are observed. Overall, DS is about half of the common chromosomal problems we find with the proportion increasing from about 1/3 at age 20 to 2/3 by age 40.
- For DS screening, low maternal serum alpha-fetoprotein was developed in the 1980’s. Second trimester screening now has evolved into the "Quad" test including AFP, human Chorionic gonadotropin (hCG), estriol (E3), and inhibin. Collectively the detection rate is about 60-65%, i.e. - if a woman is carrying a baby with DS, the test will indicate this about 65% of the time.
- DS screening moved into the first trimester beginning in the 1990’s using both serum (free β hCG and PAPP-A) as well as the ultrasound nuchal translucency (NT) described below. Together 1st trimester screening can identify about 80-85% of DS cases.
- Non-invasive prenatal screening (NIPS) has been attempted in one iteration or another for over 30 years – first looking for fetal cells in the maternal blood, which did not work satisfactorily, and then for cell free fetal DNA (cffDNA). After many false starts, several papers with in the last several years finally began showing very promising results with up to a 99% detection rate for DS. Trisomy 18 and 13 statistics are not as good. Clinical utilization in the USA has skyrocketed, but there has been under appreciation that while cffDNA is a very good screening for DS, but it is incapable of looking for many other disorders which for now can only be identified by CVS or amniocentesis and requires obtaining a direct fetal tissue specimen. Nationally there has been a significant drop in women having diagnostic procedures – instead they are relying upon these screening results. Unfortunately, we have shown that there will be many babies born with serious conditions that could have been detected by microarray that cannot be found by cffDNA. In 2018 we published a major paper in the journal Prenatal Diagnosis entitled: “The epidemic of abnormal copy number variants missed because of reliance upon noninvasive prenatal screening.” Furthermore, the chance of finding an unexpected abnormality on microarray analysis is at least 10 times as high as having a procedure complication by looking for it
- CffDNA was originally very expensive and for low risk patients – particularly whose insurance is a Medicaid plan can represent a very significant proportion of overall pregnancy costs. We have shown that the cost of detecting a case of DS by cffDNA that would be missed by combined screening approaches $1 million, which from a public health perspective was too expensive for routine use. As the cost of NIPT came down, the economics of using NIPT as a primary screening test have clearly improved, but it still misses many disorders that can only be found by diagnostic testing.
- It is very important for everyone to remember for any of the three methods above, that these are SCREENING tests and do not give a definitive answer even when abnormal. They need to be followed up by CVS or amniocentesis for a specific diagnosis. Also, as a screening test, the chance that a "positive" test really represents an abnormality is directly proportional to the underlying risk. In other words, for younger patients the chance that an abnormal test result actually represents a true abnormality is much lower than for a higher risk patient.
- It is important for all patients to remember, however, that screening results do not give a definitive answer but merely alter odds. Sometimes the laboratory report calculates a risk of nearly 1/10,000. This is for DS ONLY. Publications by Pergament and Cuckle have shown that whatever the DS risk may be on screening, the risk of other issues diagnosable by routine karyotype such as sex chromosome abnormalities, rings, translocations, and inversions can be made no lower than about 1/500. The current data show that with microarrays described under the Laboratory testing section minimum risk number is actually over 1/100. Therefore, when we counsel patients, we believe that the lowest possible risk internalized by patients in their decision-making process should be 1/100 even if the computed Down syndrome risk is reported as lower than that.
Evans et al. EPIDEMIC OF ABNORMALITIES MISSED BECAUSE OF NIPT.
Carrier Frequencies and Detection of Common Genetic Disorders
Cystic Fibrosis is perhaps the best-known Mendelian disorder in the European population. Its incidence and the component markers that cause the disease vary considerably by ethnic group. Over the past 25 years since the CF gene was identified, our understanding of the number of mutations in the gene that can cause CF has gone from literally a "few" causing most of the cases in certain groups (such as Ashkenazi Jews) to now recognizing that there are over 2000 known CF mutations. Routine CF panels have varied from about 32 markers to over 500. Now with sequencing techniques, we know of over 2000 mutations causing CF. Still the detection frequency does vary by patient’s ethnic group.
Cystic Fibrosis 2023
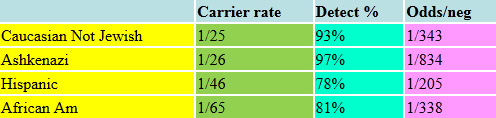
The "Ashkenazi Jewish Panel"
Advances in genetics have led to the extensive expansion of carrier screening for individuals of Ashkenazi Jewish ancestry. For decades, screening was available only for Tay-Sachs disease. The number of Ashkenazi disorders available for screening expanded during the last decade and now includes some rare disorders which area in fact no more common in the Ashkenazi population than other ethnic groups. We believe it is best to cast a “wider net,” but if couples wish to restrict their investigation to just the ethnically associated disorders, it can be done that way. Screening is now available for over 30 such conditions, including:
- Three progressive neurological conditions that begin in infancy: Tay-Sachs disease, Canavan disease, and Niemann-Pick disease, which are all fatal in early childhood
- Gaucher type 1, a chronic blood disorder resulting in anemia with neurological damage
- Fanconi anemia, a childhood illness of poor growth and predisposition to some cancers
- Bloom syndrome, a disease that affects the "automatic" and sensory functions of the body
- Familial dysautonomia
- Mucolipidosis Type 4 (ML4)
TAY SACHS DISEASE: The "classic" Ashkenazi Jewish disorder which is characterized by normal development until about 4 -6 months of age followed by progressive neurodegenerative findings including blindness, seizures, and unresponsiveness. Death is common by age 4.
CANAVAN DISEASE: Normal development up until age 2 – 4 months followed by progressive degeneration, seizures, and blindness. Lifespan can vary from early childhood into early teens.
CYSTIC FIBROSIS: Inability to transport chloride across cellular membranes leads to thick mucus in lungs and GI tract resulting in multiple infections. Lifespan has extended dramatically in the past 25 years to now average near 40.
FAMILIAL DYSAUTONOMIA: Abnormalities of autonomic and sensory nervous system affect control of body temperature, blood pressure, stress, and sensitivity to pain. Poor growth is common, and lifespan is limited.
FANCONI ANEMIA TYPE C: Short stature, bone marrow failure and a predisposition to leukemia and cancers. Shortened lifespan.
GAUCHER DISEASE Type 1: Variable presentation including spleen enlargement, anemia, fatigue, bruising, and bone deterioration.
GLYCOGEN STORAGE DISEASE TYPE 1a: Metabolic disorder characterized by hypoglycemia (sudden drops in blood sugar levels), poor growth, enlarged liver, and anemia.
MAPLE SYRUP URINE DISEASE: Abnormality of protein metabolism. Untreated symptoms include poor feeding, lethargy, seizures, and coma.
BLOOM SYNDROME: Short stature, skin rash, chromosomal breakage, infections, and high incidence of leukemia.
DIHYDROLIPOAMIDE DEHYDROGENASE DEFICIENCY: Metabolic disorder resulting in blindness, seizures, enlarged liver, and short lifespan.
FAMILIAL HYPERINSULINISM: Low levels of blood sugar resulting in seizures, poor muscle tone, sleep disorders, and poor feeding.
MUCOLIPIDOSIS IV: Progressive neurological disorder leading to muscle weakness, impaired intellect, limited vision, and shortened lifespan.
NEMALINE MYOPATHY: Low muscle tone, poor breast feeding and problems breathing. Delayed development.
USHER SYNDROME 1F: Profound hearing loss and adolescent onset of retinitis pigmentosa with severely impaired vision.
USHER SYNDROME III: Progressive hearing and vision loss. Blindness by adulthood.
JOUBERT SYNDROME: Brain abnormality produces poor muscle tone, rapid breathing, abnormal eye movements, developmental delay, and poor gait.
WALKER WARBURG SYNDROME: Congenital muscular dystrophy including multiple brain abnormalities and eye abnormalities.
Many of these disorders are very rare and some are just as common in other ethnic groups. As many of these tests are expensive and rare, the decision to screen for them individually is today not cost-effective. The carrier frequencies and detection rates of these conditions are listed in the following tables.
Ashkenazi Panel 2023
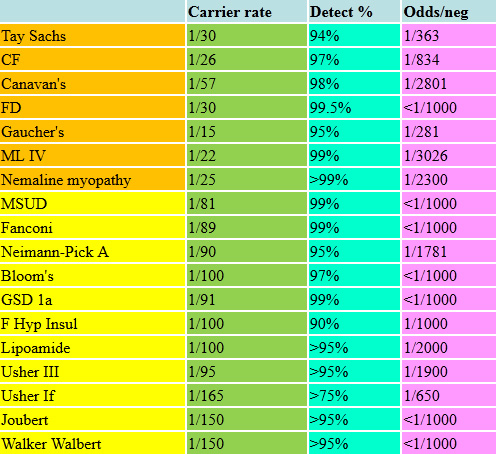
The above list is just a sample of an ever-increasing number of significant disorders for which we can now screen and test as appropriate. There are many other metabolic disorders, such as maple syrup urine disease (MSUD), an organic aciduria with severe neurological outcomes and mental retardation, and Usher syndrome, which includes profound hearing loss, vestibular abnormalities and retinitis pigmentosa resulting in blindness. Nemaline myopathy is a collection of disorders characterized by severe neuromuscular disease, weakness and hypotonia, and can be associated with early death. Familial hyperinsulinism causes hypoglycemia, which can be lethal or cause irreversible neurological damage. The make-up of the targeted panels we offer is constantly changing, so any list we show might is by definition "out of date." The carrier frequencies and detection rates of these conditions are listed in the following tables. The pan-ethnic panels are likewise expanding and improving their detection rates. For couples who have previously had screening, we check as to when it was done and by what methods as even over the past few years enough advances have occurred that would suggest a more modern panel be run.
Fragile X Syndrome
Fragile X is the most common cause of inherited mental retardation/intellectual disability, occurring in about 1/4,000 overall, but some populations are higher. It is an X- linked dominant disorder, which means that the full manifestations of the syndrome are seen in males who inherit the gene from their mothers. Its inheritance is also "imprinted," meaning that it matters from which parent the gene came—in this case, the mother. The disease is related to the number of "repeats" of a tri-nucleotide of DNA base pairs - in this case the DNA nucleotides "CGG" at the end of the FMR1 gene on the X chromosome. Classically 200 such repeats are needed for the disease to occur, and most normal women have fewer than 44 repeats on each of their X chromosomes. As the number of repeats goes up, infertility increases as does the likelihood of expansion of the number of repeats up to problem levels. Women who carry the gene may have some minor manifestations, but these are commonly unnoticed. In recent years, however, there has been an emerging appreciation of partial effects of Fragile X even with less than the usually required number of repeats needed for the full diagnosis.
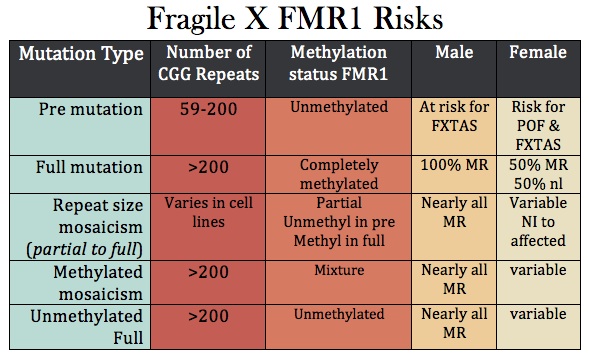
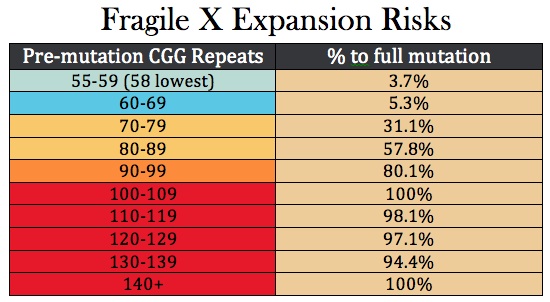
Spinal Muscular Atrophy (SMA)
Spinal muscular atrophy (SMA), which in older literature was known as Werdnig-Hoffman disease, is a rare cause of a lethal disorder with an incidence of about 1 in 4000. It is a degenerative nervous disorder with progressive downhill course. Death is common by age 2. It is autosomal recessive meaning that both parents need to be carriers, and the age of the parents is irrelevant. Carrier testing for SMA become common in big metropolitan areas such as New York about a decade ago, and it is part of the routine pan-ethnic panels. There have been recent attempts at treatment of SMA. These are potentially exciting, but a tremendous amount of work and experience are needed before they can be considered standard.
Subnavigation
Evans Pioneer Award Lecture

-
Quick Contact Info
Dr. Mark I. Evans (MD PLLC)
Phone: 212.288.1422
Fax: 212.879.2606
Email: Evans@CompreGen.com
131 E 65TH ST
NEW YORK NY 10065